Методы качественного и количественного анализа почв, растений
И удобрений
Наряду с биологическими методами исследования агрохимия широко использует лабораторные методы анализа растений, почв и удобрений. Эти методы делятся на химические, физические, физико-химические и др. Агрохимические лаборатории проводят массовые исследования качества кормов и растений. В кормах определяют содержание сырого протеина, жира, клетчатки, золы, кальция, фосфора, безазотистых экстрактивных веществ и др. Данные анализа кормов используются для организации рационального кормления сельскохозяйственных животных.
В полевых опытах с удобрениями в течение вегетационного периода отбирают образцы растений для изучения поступления питательных элементов по фазам роста и развития растений, для диагностики минерального питания растений, по данным которой при необходимости рассчитываются дозы удобрений для подкормки. Кроме того, в урожае определяют содержание белка, клетчатки, клейковины, аминокислот, крахмала (в клубнях картофеля), жира (в семенах масличных культур), сахаров и витаминов (в плодах и овощах) и др.
Агрохимические лаборатории проводят массовые анализы почв на содержание гумуса, подвижных форм фосфора и калия, магния, микроэлементов, определение кислотности и других показателей, которые используются для составления агрохимических карт и паспортов полей.
На все виды минеральных удобрений, выпускаемых химической промышленностью, утверждены стандарты (ГОСТ) или технические условия (ТУ). Если покупатель обнаружит, что удобрения не отвечают ГОСТ или ТУ (по содержанию питательных элементов и т.д.), он может предъявить заводам-поставщикам рекламации (претензии) с целью возмещения убытков. Функции арбитража в таких случаях выполняют областные проектно-изыскательские станции химизации.
В сельском хозяйстве огромное значение имеют местные удобрения (навоз, компост и др.), рациональное и эффективное использование которых предполагает организацию анализа их качества, определение содержания в них основных питательных элементов. В органических удобрениях определяют содержание влаги или сухого остатка, зольность, общего и аммиачного азота, фосфора, калия и другие показатели качества.
В агрохимии широко используется качественный и количественный анализ. Качественный анализ может быть выполнен «мокрым» и «сухим» способами. В первом случае исследуемое вещество предварительно растворяют в воде, если в воде оно нерастворимо – в другом растворителе (кислоте и т.д.), а чтобы обнаружить элементы или ионы, применяют растворы соответствующих реактивов. При этом используют реакции, сопровождающиеся внешними изменениями раствора (выпадением осадка, изменением окраски, выделением газа). Например, пожелтение раствора при добавлении к нему 5%-ного раствора AgNО3 указывает на то, что в растворе присутствует анион Н2РО4 — .
Примером «сухого» способа могут быть реакции окрашивания пламени некоторыми элементами (натрий – в желтый, калий – в фиолетовый и т.д.), а также разложение соединений при нагревании (с выделением газов, появлением специфического запаха).
Качественный анализ широко используется при распознавании минеральных удобрений, когда отсутствует документация на них, для проверки полноты тех или иных результатов количественного анализа (например, при отмывании осадков или отгоне аммиака).
Количественный анализ используется для определения количества отдельных элементов или их соединений (как органических, так и неорганических) в исследуемых веществах. Химические методы количественного анализа подразделяются на гравиметрический (весовой), объемный и газовый.
В гравиметрическом анализе количество определяемого компонента, соединения или элемента устанавливают точным взвешиванием. Масса определяемого компонента может быть определена непосредственно его взвешиванием после удаления всех других веществ из анализируемого материала, рассчитана по массе остатка после удаления определяемого компонента из анализируемого материала или по массе остатка известного химического состава, образующегося при осаждении определяемого соединения (элемента или иона) из испытуемого раствора в результате химического взаимодействия. Так, количество золы в растениях, торфе определяют взвешиванием после удаления из навески сухим сжиганием в муфельной печи органических веществ и прокаливания остатка. Количество влаги в растениях, удобрениях и почве определяют взвешиванием после их высушивания до постоянной массы, то есть после удаления определяемого вещества – воды.
Примером гравиметрического анализа, когда определяемый элемент или ион из раствора выпадает в осадок точно известного состава, массу которого и устанавливают, является определение фосфора методом Бетгера – Вагнера. В этом случае фосфор из растворов удобрений осаждается щелочной магнезиальной смесью в виде магний-аммоний фосфата – NH4MgPО4, после прокаливания осадка в муфельной печи образуется пирофосфат магния (Mg2P2О7) с точно известным содержанием фосфора. По массе полученного осадка рассчитывают количество фосфора в удобрении.
Объемный метод количественного анализа основан на изменении объема раствора реактива точно известной концентрации, израсходованного в химической реакции с определяемым веществом. В объемном анализе используют необратимые реакции, имеющие хорошо заметное окончание. Это изменение окраски самих реагирующих веществ или изменение окраски предварительно вводимых в небольших количествах в раствор индикаторов, которые изменяют свою окраску в момент окончания реакции, то есть при достижении точки эквивалентности.
Приведенные выше классические методы качественного и количественного химического анализа имеют низкую производительность и не могут удовлетворить потребность в анализе различных веществ и материалов. Поэтому с развитием оптической и электронной промышленности химические методы анализа веществ активно вытесняются более производительными и чувствительными физическими, физико-химическими и другими инструментальными методами.
Для определения фосфора, а в последнее время и азота, с помощью метода индофеноловой зелени широко используется фотоэлектрометрический метод, основанный на сравнении окраски испытуемых и образцовых растворов. С помощью фотоэлектроколориметров или спектрофотометров измеряется оптическая плотность раствора.
Для определения калия применяют метод пламенной фотометрии. Это один из методов эмиссионного спектрального анализа, основанный на измерении с помощью фотоэлемента интенсивности излучения, возникающего при возбуждении атомов определяемого элемента в пламени. Для определения используются различные марки пламенных фотометров. По показаниям микроамперметра сначала строится график для образцовых растворов с определенной концентрацией калия, затем по графику и показаниям микроамперметра для испытуемых вытяжек находят содержание калия в исследуемых объектах (почвах, растениях и т.д.).
Определение кальция, магния, меди, цинка и ряда других элементов проводится методом атомно-абсорбционной спектрофотометрии. Этот метод позволяет определять более 70 элементов и получил в последнее время широкое распространение. В основе атомно-абсорбционного метода лежит избирательное поглощение атомами химических элементов световой энергии. При проведении анализа этим методом анализируемый раствор вводят в пламя, под влиянием которого происходит переход вещества в атомное состояние. Далее пламя просвечивается внешним источником (газоразрядными лампами низкого давления и т.д.). Атомные пары определяемого элемента поглощают излучение стандартного источника. Нужная для измерения линия спектра выделяется монохроматором. Приемником излучения является фотоэлемент, возникающий ток измеряется микроамперметром. Сила тока зависит от концентрации элемента в исследуемом растворе.
Определение проводят на атомно-абсорбционном спектрофотометре. Содержание определяемых элементов в анализируемых пробах находят по калибровочному графику, который строят по стандартным растворам.
Для определения концентрации катионов (Н + , К + , Na + и др.) и анионов (NО3 — , Сl — и др.) в почве и растениях, а также окислительно-восстановительного состояния почвы в агрохимических исследованиях широко используется потенциометрический метод. Потенциометрия относится к экспресс-методам. Имея несложное оборудование (потенциометры-ионометры), можно быстро и с высокой точностью определить активность различных ионов в растворах и суспензиях.
Потенциометрический метод основан на измерении потенциала электродов, чувствительных к изменению концентрации (точки активности) тех или иных ионов в растворе. Существуют электроды, обладающие достаточно высокой чувствительностью к ионам водорода, натрия, калия, кальция, нитрат-иону и другим. Для измерения потенциала электродов их соединяют с электродами сравнения, потенциал которых постоянный и известен заранее. Возникающая разность потенциалов (электродвижущая сила) в полученной гальванической цепи усиливается и определяется компенсационным методом с точно известной электродвижущей силой другого источника постоянного тока.
Потенциометры для измерения активности водородных ионов (рН) называют рН-метрами, ионов калия, нитрата и других – ионометрами. В последнее время наряду с деструктивными методами получили распространение недеструктивные, при проведении которых не проводятся озоление, экстракция и т.п.
Из экспресс-методов очень перспективен метод инфракрасной спектроскопии, основанный на измерении интенсивности диффузного инфракрасного излучения поверхности измельченного материала. Используя этот метод, можно быстро и точно определить содержание азота, белка, аминокислот, клейковины, крахмала, жира, клетчатки, влажность, зольность и другие показатели качества растениеводческой продукции.
Этот метод можно использовать также для определения гумуса и других агрохимических показателей почвы. Для этого используется инфракрасные экспресс-анализаторы.
Источник
Анализ удобрений
РАСПОЗНАВАНИЕ МИНЕРАЛЬНЫХ УДОБРЕНИЙ ПО КАЧЕСТВЕННЫМ РЕАКЦИЯМ
При отсутствии паспорта на удобрение или при долгом его хранении возникает необходимость проведения качественного и количественного анализа.
По внешнему виду все минеральные удобрения делят на две группы – кристаллические и аморфные (порошковидные). Кристаллические удобрения хорошо растворимы в воде. Аморфные – слаборастворимы или нерастворимы.
Кристаллическое строение характерно для азотных удобрений (кроме цианамида кальция –CaCN2), калийных удобрений (кроме калимага K2SO4*2MgSO4 и печной золы) и сложных азотно-фосфорно-калийных удобрений – аммофоса (NH4H2PO4), диаммофоса ((NH4)2HPO4), нитрофоски. Аморфное состояние характерно для фосфорных и известковых удобрений.
Ход анализа кристаллических и аморфных удобрений различен.
I. Диагностика кристаллических удобрений
1) Испытывают поведение растертых кристаллов удобрения на раскаленном древесном угле: амидные и аммиачные удобрения плавятся с выделением аммиака; нитратные — вспыхивают и быстро сгорают: натриевая селитра – желтым пламенем, калийная селитра – фиолетовым, аммиачная селитра – вспыхивает и быстро сгорает с выделением белого дымка (аммиака).
2) Растворяют примерно 1 г удобрения в 15-20 мл дистиллированной воды. Раствор разливают в три пробирки и добавлением 10 %-ного NaOH или KOH, 5 %-ного BaCl2 и 1 %-ного AgNO3 устанавливают содержание конкретного катиона или аниона. Так, при действии щелочи на раствор удобрения, содержащего ион аммония (NH4Cl, NH4NO3, (NH4)2SO4), выделяется аммиак, который определяется по запаху :
Мочевина при реакции со щелочью характерных продуктов не дает.
Реакция с хлористым барием позволяет определить наличие в составе удобрения иона SO4 = . К раствору удобрения в пробирке прибавляют 2-3 капли BaCl2. Появление белого осадка (BaSO4), нерастворимого в слабой уксусной или соляной кислоте, подтверждает присутствие в составе удобрения иона SO4 =.
Реакция с азотнокислым серебром служит для определения иона Cl — :
к раствору удобрения добавляют 2-3 капли AgNO3 . Появление белого творожистого осадка указывает на присутствие в составе удобрения иона Cl — .
Калийные удобрения, в отличие от азотных, не сгорают и не плавятся на раскаленном древесном угле, а лишь потрескивают или остаются без изменения. При добавлении щелочи к раствору калийных удобрений характерных продуктов реакции не обнаруживается.
Диагностика калийных удобрений основывается как на внешних особенностях, так и на их химических реакциях (с AgNO3, BaCl2). Хлористый калий (KCl) обычно имеет вид белых мелких кристаллов; 40 %-ная калийная соль (смесь KCl с сильвинитом) – состоит из смеси белых, розовых, красных мелких кристаллов; сильвинит (mKCl*nNaCl) – представлен крупными кристаллами розового, белого, синего цвета; сернокислый калий (K2SO4) – состоит из мелких белых или кремовых кристаллов. Каинит (KCl*MgSO4*3H2O) и калимагнезия (K2SO4*MgSO4) дают хорошо выраженную реакцию на ион SO4 = . Но в отличие от сернокислого калия последние обнаруживают заметную реакцию и на хлор-ион (муть в растворе калимагнезии и белый творожистый осадок в растворе каинита).
II. Распознавание аморфных удобрений начинают с разделения их на две группы:
Качественный анализ удобрений первой группы начинают с реакции 10%-ным раствором HCl. Если при действии кислоты на удобрение наблюдается “вскипание” – это известковый материал. Суперфосфат, преципитат и гипс — не вскипают. Суперфосфат обычно представлен в виде порошка или гранул диаметром 1 — 4 мм. Водная суспензия суперфосфата имеет кислую реакцию, что определяется по изменению цвета синей лакмусовой бумажки в розовый. Преципитат и гипс отличают друг от друга реакцией с AgNO3. При добавлении в водный раствор преципитата 2-3 капель AgNO3 выпадает желтый осадок.
Плохо растворимые в воде удобрения второй группы различают по внешнему виду и реакции раствора (проба на лакмус).
Фосфоритная мука-это тонкий порошок землистого-серого цвета, имеющий нейтральную реакцию.
Томасшлак – имеет темно-серую окраску, щелочную реакцию среды. При действии на сухое удобрение кислоты бурно “вскипает”.
Цианамид кальция – иссиня-черный порошок с запахом керосина, имеющий щелочную реакцию среды. При добавлении соляной кислоты к сухому удобрению наблюдается бурное вскипание с образованием на стенках пробирки черных колец при оседании пены.
Калимаг состоит из гранул темно-серого цвета. Суспензия калимага дает хорошо выраженную реакцию на SO4 = -ион.
Реактивы: 1) дистиллированная вода; 2) 5 %-ный раствор ВаСl2 ;3) 1 %-ный раствор AgNО3 ; 4) 10 %-ный раствор NaOH или КОН; 5) 10 %-ный раствор НСl; 6) синяя лакмусовая бумага; 7) кусочки древесного угля.
Форма записи результатов
| № | Название удобрения, формула | Растворимость в воде | Отношение к раскаленному углю | Реакция с NaOH | Реакция с BaCl2 | Реакция с AgNO3 | Реакция с HCl | Отношение к лакмусовой бумажке |
ОПРЕДЕЛЕНИЕ АЗОТА В НИТРАТНЫХ УДОБРЕНИЯХ МЕТОДОМ ДЕВАРДА
Принцип метода: При действии сплава Деварда (50 % меди, 45 % алюминия и 5 % цинка) на азотнокислые соли в условиях щелочной среды происходит восстановление нитратов до аммиака:
Аммиак отгоняется в аппарате Кьельдаля и связывается титрованной серной кислотой в приемнике: 2NH3 + H2SO4 = (NH4)2SO4. По количеству связанной кислоты определяют количество аммиака, а по нему азота, содержащегося в анализируемом удобрении.
Ход анализа. Среднюю пробу удобрения растирают в фарфоровой ступке в мелкий порошок. Затем 2 г аммиачной селитры или 4 г натриевой или калийной селитры переносят в химический стакан, приливают около 100 мл дистиллированной воды и нагревают на электроплитке до кипения, помешивая раствор стеклянной палочкой. Горячий раствор фильтруют в мерную колбу на 250 мл, а стакан ополаскивают 4-5 раз водой, сливая промывные воды на фильтр. Охлаждают. Доводят объем раствора дистиллированной водой до метки и тщательно перемешивают.
Берут пипеткой 25 мл раствора, переносят в отгонную колбу аппарата Кьельдаля, приливают туда же 250-300 мл дистиллированной воды. В приемник этого аппарата наливают из бюретки 100 мл 0.2 н Н2SO4 и 2-3 капли индикатора Гроака, устанавливают под трубку холодильника так, чтобы конец ее был погружен в кислоту. Затем в отгонную колбу вносят 2-3 г сплава Деварда, приливают 2-3 капли фенолфталеина и 25 мл 40 %-ной щелочи, быстро закрывают пробкой с каплеуловителем. Содержимое колбы на 10 мин. оставляют без нагревания, затем пускают воду в холодильник, включают электроплитку, и жидкость отгоняют в течение трех часов. Полноту отгона определяют по красной лакмусовой бумажке: если последняя не посинела — отгон окончен.
Содержимое приемной колбы титруют 0.2 н NaOH до перехода сиреневой окраски в зеленую. Расчет содержания азота (в %) в удобрении проводят по формуле:
где V1 – объем кислоты, взятой для поглощения аммиака, мл; н1 – молярная концентрация эквивалентов серной кислоты (1/2 H2SO4), ммоль/мл; V2 – объем NaOH, пошедший на титрование непрореагировавшей с аммиаком кислоты, мл; н2 – молярная концентрация NaOH, ммоль/мл; 0.014 – молярная масса азота, г/ммоль; 100 – для выражения результата в %; р – разведение (250/25), m– навеска удобрения, г.
Реактивы: 1) 0.2 н H2SO4; 2) индикатор Гроака; 3) 40 %-ный раствор NaOH или КОН; 4) сплав Деварда; 5) красная лакмусовая бумага; 6) 0.2 н NaOH (8 г NaOH на 1 л раствора).
Форма записи результатов
| Название удобрения | Навеска, г | Разведение | H2SO4;, | NaOH | % N |
| мл | н | мл | н |
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ АЗОТА В АММИАЧНЫХ И АММИАЧНО НИТРАТНЫХ УДОБРЕНИЯХ ФОРМАЛИНОВЫМ МЕТОДОМ
Данный метод принят в качестве стандартного при определении содержания азота в аммиачных удобрениях – сернокислом аммонии, хлористом аммонии и др. В случае анализа аммиачной селитры результат удваивается, так как в этом удобрении азот входит в состав катиона и аниона.
Принцип метода. Основан на количественном связывании аммиака формалином с образованием нейтрального соединения гексаметилентетрамина (уротропина)- С6Н12N4. Реакция образования гексаметилентетрамина идет внейтральной среде. При этом выделяется соответствующая минеральная кислота в количестве, эквивалентном содержанию аммиачного азота:
По количеству образовавшейся кислоты, которую учитывают титрованием щелочью, определяют процент азота в удобрении.
Ход анализа. На технических весах берут навеску удобрения 2 г, помещают в химический стакан и растворяют примерно в 100 мл дистиллированной воды. Затем переносят в мерную колбу на 250 мл, доводят водой до метки и перемешивают. Если раствор получается мутный, его фильтруют. Берут пипеткой 25 мл раствора, переносят в коническую колбу, прибавляют 2-3 капли индикатора метилрота . Если раствор кислый (розовый цвет от индикатора), его нейтрализуют 0.1 н щелочью (не допуская ее избытка) до момента перехода розовой окраски в золотисто-желтую.
В другую коническую колбу помещают 10 мл 40 %-ного раствора формалина и также прибавляют к нему 2-3 капли метилрота. При наличии кислой реакции раствор формалина также нейтрализуют 0.1 н щелочью. Приготовленный раствор формалина вливают в нейтрализованный раствор анализируемого удобрения. Сразу же после сливания исследуемый раствор изменяет окраску в розовую вследствие выделения кислоты при реакции формалина с аммиаком.
К раствору с выделившейся минеральной кислотой прибавляют 2-3 капли фенолфталеина и титруют его 0.1 н щелочью. При титровании следует внимательно наблюдать за изменением цвета раствора. Вначале розовая окраска переходит в бледно-желтую, а затем она сменяется слабо-розовой, определяющей конец титрования.
Расчет содержания азота (в %) производится по формуле:
где V–объем NaOH, пошедший на титрование, мл; н – молярная концентрация NaOH, ммоль/мл; 0.014 – молярная масса азота, г/ммоль; m – навеска удобрения, г; р – разведение (250/25); 100 – для выражения результата в %.
Реактивы: 1) 0.1 н раствор NaOH (4 г NaOH на 1 л раствора), 2) метилрот; 3) 40 %-ный раствор формалина; 4) фенолфталеин.
Форма записи результатов
| Название удобрения | Навеска, г | Разведение | NaOH | N,% |
| мл | Н |
ОПРЕДЕЛЕНИЕ АЗОТА В МОЧЕВИНЕ ФОРМАЛИНОВЫМ МЕТОДОМ
Принцип метода. При нагревании с серной кислотой мочевина подвергается гидролизу с образованием аммиака и угольной кислоты.
Угольная кислота разлагается на СО2 и Н2О. Аммиак связывается с серной кислотой в сульфат аммония:
Избыток серной кислоты осторожно нейтрализуют щелочью. Далее ход анализа аналогичен определению азота в сульфате аммония.
Ход анализа. 1 г мочевины – CO(NH2)2 переносят в плоскодонную колбу из тугоплавкого стекла. Смывают приставшие к горлу колбы частицы небольшим количеством дистиллированной воды и добавляют 5 мл концентрированной H2SO4. Содержимое колбы кипятят в вытяжном шкафу до прекращения бурного выделения СО2, а затем до появления белых паров сернистого ангидрида. Охладив колбу, приливают в нее 50 мл дистиллированной воды и 2-3 капли метилрота. Нейтрализуют кислоту в колбе 5.0 н NaOH до перехода окраски в золотисто-желтую. Последующим добавлением по каплям 0.5 н H2SO4 добавляются появления розовой окраски. К нейтрализованному раствору прибавляют 20 мл 40 %-ного формалина и 2-4 капли фенолфталеина. Выделившуюся кислоту титруют до появления через желтую розовой окраски.
Расчет содержания азота (в %) производится по формуле:
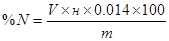
где V – объем NaOH, пошедший на титрование, мл; н – молярная концентрация NaOH, ммоль/мл; 0.014 – молярная масса азота, г/ммоль; m – навеска удобрения, г; 100 – для выражения результата в %.
Реактивы: 1) концентрированная H2SO4; 2) 5.0 н NaOH (200 г NaOH на 1 л раствора); 3) 0.5 н H2SO4 (14 мл конц. H2SO4 на 1 л раствора); 4) 40 %-ный раствор формалина; 5) 1.0 н NaOH (40 г. NaOH на 1 л раствора).
Форма записи результатов
| Название удобрения | Навеска, г | NaOH | N,% |
| мл | Н |
Определение содержания водорастворимых фосфатов в удобрениях объемным методом Шефера
Водорастворимая фосфорная кислота наиболее доступна растениям. Это, главным образом, ортофосфорная кислота и ее одно- и двузамещенные соли кальция, калия, железа и др.
Зная количество легкодоступного для растений фосфора в суперфосфате, можно правильно установить дозы фосфорных удобрений в зависимости от почвенных и климатических условий, биологических особенностей культур и фазы их развития.
Согласно стандарту, водорастворимого Р2О5 в удобрениях должно быть не менее 75 % общей фосфорной кислоты и 95 % усвояемой.
Принцип метода заключается в осаждении фосфорной кислоты раствором молибденовокислого аммония в сильнокислой среде в виде комплексного соединения (NH4)3PO4×12MoO3×2HNO3×H2O. Сильнокислая среда необходима для удержания в растворе ряда элементов. Так, концентрированная серная кислота препятствует осаждению оксида кремния. Азотная кислота предотвращает выпадение в осадок молибденовой кислоты. Кислая среда создается прибавлением смеси концентрированных серной и азотной кислот
Метод Шефера требует точного соблюдения техники анализа, так как комплексная соль может изменять свой состав при изменении условий осаждения, растворения осадка и т.п.
Образовавшийся осадок комплексной соли отмывают от примесей и растворяют в известном объеме титрованной щелочи в присутствии формалина. При растворении осадка происходит следующая реакция:
Образующийся свободный аммиак мешает дальнейшему определению, поэтому его выводят из реакции, связывая формалином в нейтральное органическое соединение гексаметилентетрамин C6H12N4.
Остаток щелочи, не пошедший на растворение осадка, оттитровывают соляной кислотой в присутствии фенолфталеина. По разности между первоначально взятым количеством щелочи, пошедшей на растворение осадка комплексной соли, которое эквивалентно содержанию фосфора в вытяжке из удобрения.
Ход анализа. Суперфосфат хорошо растирают в фарфоровой ступке. На технических весах берут навеску 2 г и переносят в мерную колбу емкостью 250 мл. Добавляют около 100 мл. Н2О и взбалтывают на ротаторе в течение 30 мин. Доводят водой до метки и после тщательного перемешивания фильтруют.
В химический стакан емкостью 100-150 мл берут пипеткой 10 мл фильтрата, прибавляют 20 мл холодной дистиллированной воды (в случае анализа простого суперфосфата), при анализе двойного суперфосфата берут 5 мл вытяжки и прибавляют 25 мл воды.
Приливают к раствору цилиндром 15 мл смеси концентрированных кислот и нагревают до появления первых пузырьков. Нельзя допускать повышения температуры выше 80 о С, так как возможно выпадение в осадок хлопьев молибденовой кислоты. Снимают стакан с огня, осторожно приливают цилиндром в середину раствора 30 мл сульфатмолибденовой жидкости. Выпавший осадок ярко-желтого цвета осторожно перемешивают круговыми движениями и оставляют на 15-18 часов. После этого производят промывание осадка. Сначала на фильтр переносят надосадочную жидкость. Осадок в стакане 4-5 раз промывают декантацией 1 %-ным раствором Na2SO4. Промывание осадка на фильтре ведут 1 %-ным раствором сульфата натрия до нейтральной реакции фильтрата (контроль по синей лакмусовой бумаге).
Отмытый осадок с фильтром помещают в колбу на 250-300 мл и приливают из бюретки 0.2 н раствор щелочи небольшими порциями до полного растворения комплексной соли. На каждые 25 мл прилитой щелочи добавляют 0.8 мл 40 % раствора формалина. Добавляют 2-3 капли фенолфталеина. Если раствор обесцветился, добавить щелочь до малиновой окраски. Избыток щелочи оттитровывают 0.1 н раствором HCl до обесцвечивания.
Расчет фосфора (в %) производится по формуле:
где V1 – объем щелочи, пошедший на растворение осадка, мл; н1 – молярная концентрация NaOH, ммоль/мл;V2–объем НСl, пошедший на титрование избытка щелочи, мл; н2– молярная концентрация НСl; ммоль/мл 0.002539 – коэффициент для перехода от количества комплексной соли к Р2О5; г/ммоль р – разведение ; m – навеска, г.
Реактивы: 1) смесь азотной и серной кислот: 30 мл Н2SO4 (d 1,84) осторожно влить в 1 л HNO3 (d 1.20). Для приготовления HNO3 ( d 1.20) –424 мл HNO3 ( d 1.41) прилить в мерную колбу объемом 1 л с 500 мл дистиллированной воды, долить водой до 1 л и перемешать; 2) раствор молибденовокислого аммония (сульфатмолибденовая жидкость): в стеклянный цилиндр, емкостью 2 л, перенести 100 г х.ч. сухого (NH4)2SO4, прилить 1 л концентрированной HNO3 (1d 1.36 – 1.37), осторожно взболтать до растворения соли. Отдельно 300 г х.ч. молибденовокислого аммония растворить в 1 л горячей дистиллированной воды, охладить до комнатной температуры и осторожно тонкой струей при непрерывном помешивании прилить в раствор азотнокислого аммония в азотной кислоте. Оставить на 48 часов. Отфильтровать через плотный бумажный фильтр. Хранить в хорошо закрытой темной склянке в темном прохладном месте; 3) 1 %-ный раствор Na2SO4 10Н2О; 4) 0.2 н раствор щелочи (KOH или NaOH); 5) 0.1 н раствор HCl (8.2 мл конц. HCl на 1 л раствора); 6) 40 %-ный раствор формалина; 7) фенолфталеин.
Форма записи результатов
| Удобрение | Навеска, г | NaOH | HCl | P2O5, % |
| мл | н | мл | н |
ОПРЕДЕЛЕНИЕ КАЛИЯ В КАЛИЙНЫХ УДОБРЕНИЯХ
Для установления дозы калийного удобрения необходимо знать процентное содержание в нем действующего вещества (К или К2О). Наиболее простой и быстрый метод определения калия – фотометрический.
Принцип метода. Фотометрия пламени – это вид эмиссионного спектрального анализа, в котором источником возбуждения излучения различных спектров служит пламя при горении смеси газов: ацетилен-воздух, пропан-кислород и др. Из-за относительно невысокой температуры пламени спектры излучения состоят из небольшого числа спектральных линий, что позволяет выделить излучения элементов при помощи светофильтров и использовать простые и имеющие сравнительно невысокую стоимость спектральные приборы – пламенные фотометры. Наиболее часто фотометрию пламени применяют при определении щелочных и щелочноземельных металлов.
Определяемые элементы поступают в плазму пламени в виде аэрозоля, полученного при распылении растворов пробы сжатым окислителем (воздух-кислород). Атомы соответствующего металла в пламени возбуждаются и дают спектр излучения пропорционально количеству атомов в растворе. При среднем содержании определяемого элементов в растворе эта зависимость линейная.
Излучение проходит через светофильтр, попадает на фотоэлемент и фиксируется гальванометром.
Ход анализа. Калийное удобрений хорошо растереть в фарфоровой ступке. На аналитических весах взять навеску удобрения 0.5 г, перенести в химический стакан. Прилить 50-100 мл холодной дистиллированной воды, размешать стеклянной палочкой до полного растворения. Отфильтровать раствор в мерную колбу емкостью 250 мл и довести водой до метки, хорошо перемешать. Взять пипеткой 10 мл раствора в другую мерную колбу емкостью 250 мл, довести водой до метки, тщательно перемешать. Последний раствор просматривают на пламенном фотометре.
Содержание в удобрении калия рассчитывают на К или К2О, используя стандартный график, построенный по шкале образцовых растворов. Расчет ведут по формуле:
где а – концентрация К2О по градуировочной кривой, мг/250 мл; m– навеска удобрения, мг; р – разведение – 250/10.
Приготовление калибровочной шкалы. 0.7915 г КСl растворяют в 1 л дистиллированной воды. В 1 мл полученного стандартного раствора содержится 0.5 мг К2О. Эталонные растворы готовят в мерных колбах емкостью 250 мл; приливая из бюретки возрастающие количества стандартного раствора: 0.5; 1; 2; 4; 5; 10; 20; 40; 50 мл. Колбы доводят до метки дистиллированной водой и перемешивают.
Приготовленные стандартные растворы, начиная с наименьшей концентрации, вводят в пламя прибора и записывают показания гальванометра. Концентрацию элемента в испытуемых растворах (в мг/250 мл) находят по градуировочным кривым, которые строят в координатах: отсчет по шкале прибора – концентрация элемента.
Шкала для приготовления эталонных растворов
| Номер колбы | |||||||||
| Стандартный раствор, мл | 0.5 | ||||||||
| Содержание К2О в мг/250 мл | 0.25 | 0.5 | 1.0 | 2.0 | 2.5 | 5.0 | 10.0 | 20.0 | 25.0 |
Форма записи результатов
| Удобрение | Навеска, г | Разбавление | Отсчет по шкале прибора | Концентрация элемента в анализируемом растворе, мг/250 мл | К2О, % |
ОПРЕДЕЛЕНИЕ ОБЩЕЙ НЕЙТРАЛИЗУЮЩЕЙ СПОСОБНОСТИ ИЗВЕСТКОВЫХ УДОБРЕНИЙ
Основной целью применения известковых удобрений является нейтрализация почвенной кислотности. Факторами нейтрализации в известковых удобрениях являются СаСО3, МgCO3, CaO, Ca(OH)2. Для агрономических целей нет необходимости определять каждое из указанных соединений отдельно. Достаточно определить их общую нейтрализующую способность.
Принцип метода. Известковое удобрение обрабатывают при нагревании титрованной соляной кислотой:
Соляную кислоту для анализа берут с избытком. Остаток ее оттитровывают щелочью: HCl + NaOH = NaCl + 2H2O
По разности между количеством HCl, взятой для анализа, и ее остатком устанавливают количество соляной кислоты, пошедшей на реакцию. Этот объем эквивалентен нейтрализующей способности суммы карбонатов, оксидов и гидроксидов кальция и магния.
Результат выражают в процентах СаО или СаСО3.
Ход анализа. 2 г тонко измельченного удобрения переносят в стакан или колбу, приливают пипеткой 200 мл 0.5 н HCl , перемешивают и ставят для нагревания на кипящую водяную баню. Нагревание ведут при помешивании стеклянной палочкой до полного растворения осадка. Затем фильтруют в сухую посуду. Берут 50 мл фильтрата в коническую колбу на 250 мл, добавляют 2-3 капли фенолфталеина и титруют несвязанную кислоту 0.5 н раствором NaOH до слабо-розовой окраски, не исчезающей в течение минуты.
Результаты вычисляют по формуле:
где н1 – молярная концентрация HCl; ммоль/мл; V2 — объем NaOH, пошедшей на титрование, мл; н2 – молярная концентрация NaOH, ммоль/мл; 0.05 – ммоль (1/2 СаСО3), г; m – навеска, отвечающая 50 мл раствора, взятого для титрования, г.
Реактивы: 1) 0.5 н раствор HCl (41 мл конц. НСl на 1 л раствора); 2) 0.5 н раствор NaOH (20 г NaOH на 1 л раствора); 3) фенолфталеин.
Форма записи результатов
| Удобрение | Навеска, г | HCl | NaOH | СаСО3,% |
| мл | н | мл | н |
Гипс применяется для химической мелиорации солонцов. Дозы его устанавливают в зависимости от содержания в почве поглощенного Na, а также от содержания CaSO4∙2H2O в материале, используемом для гипсования.
Принцип метода. Навеска гипса растворяется при нагревании в разбавленной соляной кислоте:
В растворе после отделения примесей определяют содержание сульфат-ионов, которые осаждают хлористым барием:
Осадок BaSO4 прокаливают и взвешивают. Зная содержание в исследуемом материале ионов SO4 2- , можно рассчитать и содержание в нем гипса.
Ход анализа. Навеску гипса около 1 г берут на аналитических весах, переносят в фарфоровую чашку и прибавляют 50 мл разведенной HCl (реактив 1). Чашку накрывают часовым стеклом и ставят на электроплитку. Нагревание раствора ведут постепенно до кипения. Затем снимают стекло и смывают его в чашку дистиллированной водой, содержимое чашки выпаривают на водяной бане досуха. Сухой остаток смачивают 5-10 мл разведенной HCl (реактив 1), приливают туда 50 мл горячей дистиллированной воды и хорошо размешивают стеклянной палочкой. Далее для выделения выпавшего остатка кремнекислоты, песка, глины и других примесей полученный раствор фильтруют через беззольный фильтр в мерную колбу емкостью 100 мл. Осадок на фильтре промывают горячей дистиллированной водой, подкисленной HCl (реактив 3).
По охлаждении раствор в колбочке доливают до метки дистиллированной водой, закрывают пробкой и взбалтывают. В химический стакан берут пипеткой 25 мл раствора для осаждения иона SO4 2- и нагревают до кипения. Одновременно в стеклянной пробирке доводят до кипения 5 мл 10 %-ного раствора BaCl2 (реактив 2) и приливают его в стакан с кипящим раствором. Чтобы добиться полноты осаждения SO4 2- -ионов и образования более крупных кристаллов BaSO4 стакан накрывают стеклом и оставляют на 4 часа.
Пока идет осаждение, готовят чистый, прокаленный до постоянного веса фарфоровый тигель. По истечении 4 часов содержимое стакана фильтруют через плотный фильтр (синяя лента), промытый предварительно кипящей дистиллированной водой, подкисленной HCl (реактив 3), для удаления из фильтровальной бумаги следов сульфат-ионов.
Поскольку мелкокристаллический осадок BaSO4 способен проходить даже через плотный фильтр, следует колбу, в которую собирается фильтрат, ставить на черную бумагу. Это позволяет легко обнаружить “проскочивший” осадок. Если это произошло, фильтрат подкисляют соляной кислотой (реактив 4), нагревают до кипения и вновь фильтруют.
Когда на фильтр перенесен весь осадок из стакана, последний многократно обрабатывают реактивом 3, сливая промывные воды на фильтр. Промывание осадка продолжают до тех пор, пока в промывной жидкости будет отсутствовать реакция на Ва 2+ (с 10 %-ной H2SO4). Отмытый фильтр с осадком переносят в фарфоровый тигель и подсушивают на электроплитке. Затем тигель переносят в муфель для прокаливания осадка. Температура в муфеле не должна подниматься выше 750 о С, т.к. при 800 о С BaSO4 уже разлагается. По этой же причине прокаливание не следует излишне затягивать (после озоления достаточно 20-минутного прокаливания).
Тигель с осадком охлаждают в эксикаторе и взвешивают на аналитических весах, доводя до постоянного веса.
Результат анализа выражают в % к удобрению по следующей формуле:
где а – масса тигля с осадком после прокаливания, г; б – масса пустого тигля, г; в – масса золы фильтра, г; 0.4114 – граммы SO4 2- , отвечающие 1 г BaSO4; 1.7922 – граммы CaSO4∙2H2O, отвечающие 1 г SO4 2- ; m– навеска удобрения, г; р – разведение (100/25).
Реактивы: 1) HCl разведенная (1 объем HCl (d 1.19) приливают к 3 объемам дистиллированной воды); 2) BaCl2, 10 %-ный раствор; 3) дистиллированная вода, подкисленная HCl (2-3 мл HCl (d 1.29) на 500 мл воды); 4) HCl, 10 %-ный раствор (236,4 мл конц. HCl на 1 л раствора); 5) H2SO4 – 10 %-ный раствор (60,6 мл конц. . H2SO4 на 1 л раствора).
Форма записи результатов
| Удобрения | Навеска, г | Разведение | Масса пустого тигля, г | Масса золы фильтра, г | Масса тигля с прокаленным осадком, г | CaSO4∙2H2O,% |
АНАЛИЗ ОРГАНИЧЕСКИХ УДОБРЕНИЙ
Определение питательных веществ в органических удобрениях используется для оценки их качества, а также для расчета доз внесения удобрений в почву.
ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ АЗОТА В НАВОЗЕ
Принцип метода. Органическое вещество навоза озоляют концентрированной серной кислотой с добавлением селена (катализатор). Серная кислота при температуре кипения в присутствии органических веществ распадается на сернистый газ, воду и кислород:
Кислород, выделяющийся при распаде серной кислоты, окисляет аминокислоты. При этом выделяется аммиак, углекислый газ и органическая кислота, которая в дальнейшем тоже окисляется до воды и углекислого газа. Аммиак связывается серной кислотой в сульфат аммония: 2NH3 + H2SO4 = (NH4)2SO4, и отгоняется из щелочного раствора в аппарате Кьельдаля::
Аммиак по трубке холодильника попадает в приемник с титрованной серной кислотой и связывается ею с образованием вновь сульфата аммония:
По количеству кислоты, связанной выделившимся аммиаком, определяют процент азота в навозе.
Ход анализа. 5 г (1 г сухого) навоза помещают в колбу Кьельдаля . Заливают навеску 20 мл концентрированной H2SO4 и дают постоять 30 минут (лучше оставить до следующего занятия), чтобы органическое вещество навоза частично обуглилось. Затем вносят в колбу 0.1 г селена. Для полного разрушения органического вещества необходима температура около 400 о С, дальнейшее повышение температуры ведет к потере азота, поэтому для поддержания температуры на требуемом уровне добавляют в колбу 5 г К2SO4. После этого содержимое колбы нагревают до полного обесцвечивания. Далее колбу охлаждают, осторожно приливают немного дистиллированной воды и количественно переносят в отгонную колбу, доводя объем жидкости в отгонной колбе до 300 мл. Прибавляют 2-3 капли фенолфталеина. В приемник аппарата наливают из бюретки 50 мл 0.1 н Н2SO4 и 2-3 капли индикатора Гроака, устанавливают под трубку холодильника так, чтобы ее конец был погружен в кислоту. После этого в отгонную колбу добавляют 80 мл 40-50 % NaOH и быстро закрывают пробкой. Пускают воду в холодильник, включают электроплитки и кипятят до тех пор, пока не перегонится 2/3 объема жидкости. Полноту отгона проверяют по красной лакмусовой бумажке.
Остаток серной кислоты в приемнике оттитровывают 0.1 н щелочью.
Расчет (в %) производится по формуле:
где V1 – объем Н2SO4, взятой в приемную колбу для поглощения аммиака, мл; н1 – молярная концентрация эквивалентов серной кислоты (1/2 H2SO4), ммоль/мл; V2 – объем NaOH, пошедший на титрование непрореагировавшей с аммиаком кислоты, мл; н2 – молярная концентрация NaOH, ммоль/мл; 0.014 – молярная масса азота, г/ммоль; 100 – для выражения результата в %; m – навеска удобрения, г.
Реактивы: 1) концентрированная Н2SO4; 2) 40 % раствор NaOH; 3) 0.1 н Н2SO4 (2,8 мл конц. Н2SO4 на 1 л раствора); 4) фенолфталеин; 5) индикатор Гроака; 6) красная лакмусовая бумага; 7) 0.1 н NaOH (4 г NaOH на 1 л раствора)ё.
Форма записи результатов
| Навеска, г | Объем Н2SO4, взятый для озоления, мл | Н2SO4 | NaOH | % N |
| мл | н | мл | н |
ОПРЕДЕЛЕНИЕ АММИАЧНОГО АЗОТА В НАВОЗЕ ПО И.РОМАШКЕВИЧУ КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
Принцип метода. Аммиак вытесняется из навоза и одновременно связывается 0.05 н соляной кислотой (NH3 +HCl = NH4Cl). В полученной вытяжке производится колориметрическое определение NH 4+ , основанное на взаимодействии солей аммония с реактивом Несслера с образованием комплексной соли желтого цвета (йодистого меркураммония):
Присутствующие в растворе катионы кальция, магния и др. ионы с реактивом Несслера дают нерастворимые осадки и вызывают помутнение раствора. Поэтому к испытуемому раствору добавляют сегнетовую соль (калий-натрий вин
нокислый, KOOC(CHOH)2COONa*4H2O), которая связывает ионы в недиссоциирующие соединения
Ход анализа: На технических весах берут навеску навоза 10 г и переносят в 500 мл колбу с широким горлом. Туда же приливают 250 мл 0.05 н HCl и встряхивают на ротаторе в течение 30 минут. Затем содержимое колбы фильтруют. Для получения прозрачного фильтрата необходимо сразу перенести как можно больше твердых частиц навоза. Из отфильтрованной вытяжки берут 5 мл в мерную колбу на 100 мл, прибавляют 4 мл 25 %-ного раствора сегнетовой соли, доводят общий объем жидкости водой до 80-90 мл, приливают 2 мл реактива Несслера и доводят до черты. Хорошо перемешивают.
Одновременно готовят образцовые растворы. В мерные колбы на 100 мл помещают 1; 2; 5; 8; 10; 15 мл рабочего раствора хлористого аммония, приливают дистиллированной воды, 4 мл сегнетовой соли и 2 мл реактива Несслера, доводят объем до метки. Хорошо перемешивают. Через 15 минут растворы просматривают на ФЭКе при синем светофильтре.
Содержание аммонийного азота (в %) рассчитывают по формуле:
где а –количество азота по графику, мг/100 мл; V – общий объем раствора, мл ; V1 – объем раствора, взятый для окрашивания, мл ; m – навеска, г; 1000 – перевод мг в г.
Реактивы. 1) 0.05 н HCl (4.1 мл конц HCl на 1 л раствора); 2) 25 %-ный раствор сегнетовой соли; 3) реактив Несслера; 4) образцовый раствор NH4Cl: 0.7405 х.ч. NH4Cl растворяют в дистиллированной воде и доводят объем до 1 л. Затем 20 мл этого раствора переносят в мерную колбу и вновь доводят до 1 л. В 1 мл последнего образцового раствора содержится 0.005 мг NH4 + .
Все реактивы готовят на дистиллированной воде, не содержащей аммиака.
| Количество образцового раствора, мл | ||||||
| Концентрация образцового раствора, мг/100 мл | 0.005 | 0.01 | 0.025 | 0.04 | 0.05 | 0.075 |
| Отсчет по ФЭКу |
| Удобрение | Навеска, г | Отсчет по ФЭКу, D | NH4 + (по графику, мг/100 мл) | % N |
ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ ФОСФОРА В НАВОЗЕ
Для определения содержания фосфора в органических удобрениях широко используется метод, предложенный Дениже.
Принцип метода. Навеску вещества озоляют сухим способом. В растворе фосфор определяют колориметрически.
Основой данного метода является способность фосфат-иона в слабокислой среде образовывать с молибденовокислым аммонием фосфор-молибденовую гетерополикислоту H7(P(Mo2O7)6)*nH2O. При добавлении сильного восстановителя – хлористого олова – шестивалентный молибден частично восстанавливается до пятивалентного, при этом образуется фосформолибденовая синь и раствор окрашивается в голубой цвет. Поскольку состав получаемого окрашенного комплекса зависит от кислотности среды, концентрации молибдата аммония и свойств восстановителя, необходимо строго соблюдать указания используемой методики.
Ход анализа. Навеску навоза (около 1 г) помещают в прокаленный тарированный тигель и подвергают сухому озолению в муфельной печи. Температура не должна превышать 500-525 о С. После 30-минутного прокаливания тигли вынимают и охлаждают на воздухе. Прибавляют 5-6- капель концентрированной HNO3 или 30 %-ного пергидроля, затем после испарения окислителя тигель снова ставят в муфель на 30 мин. Если полного сжигания не произошло, в охлажденные тигли вновь добавляют окислитель, выпаривают и прокаливают Осторожно переносят тигли в эксикатор, охлаждают и взвешивают на аналитических весах. Озоление считается законченным, если разница двух последних взвешиваний не превышает ±0.0005 г.
Полученную золу в тигле увлажняют несколькими каплями дистиллированной воды. Приливают 5 мл 25 %-ного раствора HCl и тщательно размешивают стеклянной палочкой (работа ведется в вытяжном шкафу). Для более полного растворения золы и снижения концентрации раствора в тигель приливают 15-20 мл горячей дистиллированной воды. Растворенную золу вместе с нерастворившимися частицами переносят через воронку в мерную колбу емкостью 100 мл (колба № 1), многократно промывая тигель и палочку дистиллированной водой и сливая промывную жидкость в ту же колбу. Охлажденный раствор доводят до метки, закрывают чистой пробкой и перемешивают.
После того как нерастворившиеся частицы осядут на дно колбы, осторожно пипеткой берут 10 мл прозрачного раствора и переносят в другую мерную колбу на 100 мл (колба № 2). Сюда же добавляют 1-2 капли фенолфталеина и нейтрализуют 1%-ным раствором аммиака до слабо-розовой окраски. Нейтрализованный раствор доводят до метки и хорошо перемешивают. 10 мл раствора из колбы № 2 переносят в третью мерную колбу емкостью 100 мл, приливают 10 мл 27 %-ного раствора серной кислоты, 10 мл 2 %-ного раствора молибденовокислого аммония и 60 мл дистиллированной воды. Содержимое колбы доводят до метки и хорошо перемешивают. Добавляют 7 капель хлористого олова и снова перемешивают. Через 5 мин. раствор колориметрируют (светофильтр красный).
Приготовление калибровочной шкалы. 0.1917 к КН2РО4 растворяют в дистиллированной воде, переносят в мерную литровую колбу и доводят до метки. Для приготовления рабочего раствора берут пипеткой 20 мл основного раствора и разбавляют в литровой колбе водой до метки. Получают рабочий раствор с концентрацией Р2О5 0.002 мг/мл.
Для построения калибровочного графика готовят следующие серии растворов:
| Рабочий раствор, мл | ||||||||||
| Концентрация Р2О5, мг\100 мл | 0.01 | 0.02 | 0.03 | 0.04 | 0.05 | 0.06 | 0.07 | 0.08 | 0.09 | 0.10 |
| Отсчет по ФЭКу, D |
Окрашивают эталонные растворы так же, как и анализируемые (за исключением нейтрализации по фенолфталеину).
Содержание Р2О5 (в %) рассчитывают по формуле:
где а- количество Р2О5 по графику, мг/100 мл; р – разведение; 100 – для выражения данных в %; m – навеска, г;1000 – для перевода мг Р2О5 в г.
Реактивы: 1) НСl, 25 %-ный раствор (63,4 мл конц. НСl на 100 мл раствора); 2) NH4OH, 1 %-ный раствор (43,7 мл конц. NH4OH на 1 л раствора) ; 3) H2SO4, 27 %-ный раствор (184,4 мл конц. H2SO4 на 1 л раствора); 4) молибденовокислый аммоний, 2 %-ный раствор; 5) раствор хлористого олова (берут 2.5 г SnCl2∙2H2O и растворяют при нагревании в 24 мл НСl (d 1.19). После охлаждения приливают дистиллированную воду до 100 мл и получают раствор олова в 10 %-ном растворе НСl). В работе следует использовать свежеприготовленный раствор двухлористого олова; 6) фенолфталеин.
Форма записи результатов
| Удобрение | Навеска, г | Разведение | Отсчет по ФЭКу, D | Р2О5 (по графику, мг/100 мл) | Р2О5, % |
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
1. Петербургский А.В. Практикум по агрономической химии. – М., 1968.
2. Практикум по агрохимии / Под ред. В.Г.Минеева. – М.,1989.
3. Практикум по агрохимии /Под ред. А.С.Радова. – М., 1985.
4. Химический анализ почв: Учеб. пособие / Растворова О.Г., Андреев Д.П., Гагарина Э.И. и др. — СПб., 1995.
5. Воробьева Л.А. Химический анализ почв. М., 1998.
РАСПОЗНАВАНИЕ МИНЕРАЛЬНЫХ УДОБРЕНИЙ ПО КАЧЕСТВЕННЫМ РЕАКЦИЯМ. 2
ОПРЕДЕЛЕНИЕ АЗОТА В НИТРАТНЫХ УДОБРЕНИЯХ МЕТОДОМ ДЕВАРДА. 4
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ АЗОТА В АММИАЧНЫХ И АММИАЧНО-НИТРАТНЫХ УДОБРЕНИЯХ ФОРМАЛИНОВЫМ МЕТОДОМ. 7
ОПРЕДЕЛЕНИЕ АЗОТА В МОЧЕВИНЕ ФОРМАЛИНОВЫМ МЕТОДОМ. 6
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВОДНОРАСТВОРИМЫХ ФОСФАТОВ В УДОБРЕНИЯХ ОБЪЕМНЫМ МЕТОДОМ ШЕФЕРА. 7
ОПРЕДЕЛЕНИЕ КАЛИЯ В КАЛИЙНЫХ УДОБРЕНИЯХ. 9
ОПРЕДЕЛЕНИЕ ОБЩЕЙ НЕЙТРАЛИЗУЮЩЕЙ СПОСОБНОСТИ ИЗВЕСТКОВЫХ УДОБРЕНИЙ. 11
АНАЛИЗ ГИПСА. 12
ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ АЗОТА В НАВОЗЕ. 14
ОПРЕДЕЛЕНИЕ АММИАЧНОГО АЗОТА В НАВОЗЕ ПО И.РОМАШКЕВИЧУ КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ 15
ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ ФОСФОРА В НАВОЗЕ. 17
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ. 19
Составители: Брехова Любовь Ивановна
Стахурлова Лариса Дмитриевна
Редактор Бунина Т.Д.
Дата добавления: 2014-12-09 ; просмотров: 763 ; Нарушение авторских прав
Источник